




Introduction
Glycation is a non enzymatic reaction or Maillard reaction between reducing sugars and proteins (McCance et al., 1993). Advanced glycation end-products (AGEs) from non-enzymatic glycation are believed to participate in the pathogenesis of such microvascular complications as nephropathy, arteriosclerosis, retinopathy, neuropathy, and cataracts in the aged population and people with diabetes mellitus (Vlassara et al., 1994; Brownlee, 2001; Ahmad et al., 2007).
The non-enzymatic glycation can be separated into three main stages: early, middle and late stage (Lapolla et al., 2005). The early stage leads to the formation of reversibly glycosylated proteins (Schiff base) that subsequently rearrange into stable Amadori products. This reaction is non-enzymatic. Formation of the Schiff base occurs over a period of several hours, whereas formation of Amadori products takes days (Brownlee et al., 1988; Lapolla et al., 2005). These reactions are reversible, and the equilibrium is highly dependent on substrate concentrations and incubation time (Bonnefont-Rousselot, 2002). At the middle stage, the Amadori product degrades through an oxidation and dehydration reaction into a variety of dicarbonyl intermediates such as glyoxal, methylglyoxal and deoxyglucosones, which are much more reactive than sugars and react again with free amino groups of proteins (Thornalley, 1996; Lapolla et al., 2005). Methylglyoxal (MGO) is a very reactive intermediate, formed by anaerobic decomposition of triose phosphate intermediates in glycolysis. At the late stage, the products undergo conversion to dicarbonyl intermediates to form advanced glycation end-products (AGEs) (Brownlee et al., 1988).
Aminoguanidine (AG) is a well-known inhibitor of the formation of AGEs. Among various inhibitors of advanced protein glycation, AG is one of the most promising compounds (Bucala et al., 1995). AG, a nucleophilic hydrazine, reacts with Amadori fragmentation products (Edelstein & Brownlee, 1992). Its beneficial effect has been demonstrated in vitro and in vivo in animal models, with a significant reduction in the AGE levels in blood and tissues (Soulis et al., 1996; Kelly et al., 2001). AG significantly has decreased the formation of AGE-hemoglobin, and was effective in inhibition of the inducible form of NO-synthase. However, administration of AG in therapy is necessary to consider carefully because of its possible toxic and prooxidative effects (Ou and Wolff, 1993).
Therefore, screening of novel AGE inhibitors from natural sources seems feasible. Diet rich in fruit and vegetables protects against degenerative diseases due to the presence of bioactive substances that exert specific actions on biological targets, including anti-glycation activity (Bousova et al., 2005; Ardestani & Yazdanparast, 2007a; Hsieh et al., 2007). Compounds with antioxidant capacity such as polyphenols have been proven to exert anti-glycation effects at physiological concentrations (Kim & Kim, 2003; Lunceford & Gugliucci, 2005; Rudnicki et al., 2007).
Therefore, the plants might offer a new source of glycation inhibition agents. So far, there have been no reports on the inhibition of protein glycation by the extracts from Aloe vera leaf, Aloe arborescens leaf, Aloe vera callus, Portulaca oleracea and cacao (Theobroma cacao L.) bean husk. This study aimed to evaluate and compare the anti-glycation capacities of extracts from these five materials with that of AG as a positive control. Correlation between the contents of phenolics and flavonoids and anti-glycation activity was also analysed to identify the components responsible for the activity.
Materials and Methods
Aluminum chloride was purchased from Jusei Chemical Co., Ltd. (Tokyo, Japan). All reagents, unless otherwise stated, were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Cacao bean husk (CBH) was supplied by Lotte Groups R&D Center (Seoul, Korea); Aloe vera and Aloe arborescens leaves were supplied by KJM Aloe Co. Ltd. (Seoul, Korea). Aloe vera callus at maintained at New Biomaterials Lab of Kangwon National University (Chuncheon, Korea). Portulaca oleracea was purchased from a local market (Herbal Love, Seoul, Korea). CBH was supplied by Lotte Group R&D Center (Seoul, Korea) and defatted by extraction with ethyl ether in a Soxhlet apparatus for 12 h. All raw materials were dried and grinded to 80 mesh.
Samples were extracted with hot water, cold water or 60% EtOH solution. Additionally, Aloe vera leaves were extracted with 60% acetone (v/v), 95% acetone (v/v), hexane, chloroform, chloroform : EtOH (3:1, v/v) or 95% EtOH.
Briefly, powdered samples (30 g) were extracted twice for 2 h with 300mL of cold (30 ℃) or hot water (95 ℃), and centrifuged (5000 x g, 15min). The supernatants were concentrated in a rotary vacuum evaporator (N-N Series, EYELA Co., Tokyo, Japan) and evaporated to dryness under vacuum. Powdered samples were extracted with solvents at a ratio of 60 mL/g for 3 h at room temperature three times, followed by filtration through filter paper (Whatman No. 4, Springfield Mill, Huddersfield, UK). The filtrates were combined and concentrated in a rotary vacuum evaporator until dryness. Samples were kept in a desiccator until use. Depending on the assay, the extracts that were insoluble in appropriate buffer were first dissolved in methanol and then diluted to different concentrations needed for a particular assay.
Dot-blot DPPH staining was used to asses rapid screening of antioxidation, and was determined according to the method described by Soler-Rivas et al. (2000). Extracts were dissolved in water or methanol; 5-μL aliquots were applied on Merck Silica gel F254 plates and allowed to dry for a few minutes. Ascorbic acid was used as a positive control. A 0.4 mM DPPH solution in methanol was sprayed on the plates until they were evenly covered. The excess solution was removed with tissue paper. Stained silica layer revealed a purple background with spots indicating radical scavenger activity. The intensity of the yellow color depends on the amount and nature of the radical scavenger present in the sample.
DPPH radical scavenging activity of the extracts was determined using the method described by Choi et al. (2002). Test solutions were prepared by adding 1 mL of a 0.3 mM DPPH solution in ethanol to samples (2.5 mL) diluted to different concentrations. To prepare blank solutions, 1 mL of ethanol was used instead DPPH. To prepare a negative control, ethanol was used instead of samples. As DPPH is sensitive to light, solutions were protected from light as much as possible. The reactions were allowed to proceed at room temperature for 30 min. The absorbance was measured at 518 nm (Spectronic Genesys-5TM spectrophotometer, NY, USA) and converted into the percentage antioxidant activity using the following equation:
TBA-RS (thiobarbituric acid-reactive species) assay was used to assess lipid peroxidation of the extracts and was determined according to William & Du (1992). Briefly, 4.1 mL of 2.51% linoleic acid in ethanol, 8 mL of 50mM phosphate buffer (pH 7.0), and 3.9 mL of distilled water were added to 4 mL of samples (0.01-1 mg/mL). The solution was capped, thoroughly mixed and incubated for 5 days at 40 ℃ in the dark with shaking. After incubation, 1 mL of the solution was added to 2 mL of 20% trichloroacetic acid (TCA) in distilled water and 2 mL of 0.65% thiobarbituric acid (TBA; Sigma T-5500). The reaction mixture was allowed to stand at 95 ℃ for 20 min and then was cooled rapidly. After centrifugation for 10 min at 5000 x g, the absorbance of the supernatant was measured at 532 nm. The % inhibition of lipid peroxidation was calculated as (absorbance of test sample/ absorbance of control)×100.
Sample of 0.1 mL was added to 1 mL carbonate buffer (0.1 M, pH 10.8) containing 0.25 M nitroblue tetrazolium (NBT) at 37 ℃; the absorbance at 530 nm was measured at 10 and 15 min after mixing (Johnson et al., 1982)
BSA (50 mg/mL) was incubated with 100 mM MGO under sterile conditions in 0.1 M phosphate buffer (pH 7.4) at 37 ℃ for 7 days. Samples were added in the concentration range of 0.01-1 mg/mL, and fluorescence was measured at excitation and emission wavelengths of 335 and 460 nm, respectively, with a spectrofluorometer(Fluoroskan Ascent; Thermo Scientific Inc., FL, USA) (Wu & Yen, 2005). The % inhibition of AGE formation was calculated as (fluorescence of test sample/fluorescence of control)×100.
RNase (1 mg/mL) was incubated with 0.5M ribose in 0.02 M phosphate buffer (pH 7.2) at 60 ℃ for 1, 2 or 3 days. Where indicated, RNase was incubated with a chemical glycation or extracts in the presence of 0.5 M ribose. Control samples were incubated under similar conditions but without glycation inhibitor or extracts. Samples were added in the concentration range of 0.01-1 mg/mL. Fluorescence was measured at excitation and emission wavelengths of 335 and 460 nm, respectively (Fatima et al., 2008). The % inhibition of AGE formation was calculated as (fluorescence of test sample/ fluorescence of control)×100.
Total phenolics in the extracts were determined using the Folin-Ciocalteu reagent method (Bray & Thorpe, 1954). To 0.2 mL of each extract, 2 mL of 2% sodium carbonate solution was added. After 2 min, 0.2 mL of 1 N Folin-Ciocalteu reagent was added and the mixture was allowed to stand for further 30 min. The absorbance was measured at 750 nm using a spectrophotometer (Spectronic Genesys-5TM spectrophotometer, NY, USA). A calibration curve using (+) catechin was obtained.
The flavonoid content was determined by the aluminum chloride method (Parthasarathy et al., 2009) using (+) catechin as a standard. Extracts and (+) catechin standard solution were prepared in ethanol (1 mg/mL) and mixed (0.25 mL) with 1.25 mL of distilled water, followed by addition of 75 μL of 5% sodium nitrite. After 6 min, 150 μL of 10% aluminum chloride was added and the mixture was allowed to stand for further 5 min, after which 0.5 mL of 1M sodium hydroxide was added. The mixture was brought to 2.5 mL with distilled water and mixed well. Blank solutions contained the same reagents, except that the extract or (+) catechin standard solution was substituted with 0.25 mL of ethanol. The absorbance was measured immediately at 510 nm using Spectronic Genesys-5TM.
Results and Discussion
Antioxidant capacity of extracts was detected by dot-blot assay, a rapid DPPH staining method using TLC method (Fig. 1). The purple area on the plate indicates no free radical scavenging activity, while the yellow areas indicate the presence of such activity. The size of the yellow zones depends on the amount of a radical scavenger present in the sample. In chloroform, hexane, 95% acetone and 95% EtOH extracts of Aloe vera leaves, cold water, hot water and 60% EtOH extracts of Portulaca oleracea and the 60% EtOH extract of CBH, the yellow color zones appeared up to a concentration of 0.12 mg/ mL. However, the natural color of the concentrated samples might interfere with the measurements: in the chloroform, chloroform : EtOH (3:1,v/v) and hexane extracts of Aloe vera leaf, the color of the concentrated spots was green and that of the CBH extract was brown. The strongest dot-blot staining was detected by eye in the 60% EtOH extracts of all samples, and especially in those of Aloe vera leaves, Portulaca oleracea and CBH.

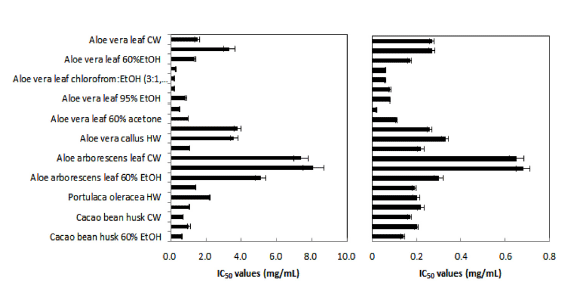
The radical scavenging and antioxidant activities of the extracts were investigated with the DPPH radical scavenging and lipid peroxidation assays, respectively, using ascorbic acid as a positive control. The results were expressed as IC50 values (Fig. 2). Of the 21 samples tested, Aloe vera leaf extracts (chloroform : EtOH and hexane) had the greatest DPPH radical scavenging activities with the same IC50 value of 0.2 mg/mL. Aloe vera leaf extracts (95% and 60% acetone) and CBH extracts (60% EtOH) also had good DPPH radical scavenging activities. High anti-lipid peroxidation activities with IC50 values of 0.06, 0.06 and 0.2 mg/mL were observed in Aloe vera leaf extracts with chloroform, chloroform: EtOH (3:1,v/ v) and 95% acetone, respectively.
The effects of the extracts on the early, middle and late stages of glycation were examined by the NBT, MGO-BSA and RNase-ribose assays, respectively. The inhibitory effects of the extracts at different concentrations (0.01, 0.1 and 1 mg/ mL) are shown in Table 1.
Among the 21 extracts, Aloe vera leaf extracts (95% EtOH and 95% acetone), Aloe vera callus extract (60% EtOH), Portulaca oleracea extracts (hot water and 60% EtOH) and CBH extracts (cold and hot water) showed slight inhibitory effects on fructosamine formation, and other tested extracts had no inhibitory effects. AG and rutin as a positive control had also no inhibitory effects on fructosamine formation, This is in line with the report by Ardestani & Yazdanparast (2007b), who found that AG had no inhibitory effect on the early stage of glycation. (+) catechin and (−) epicatechin showed a strong inhibitory rate of 59.4% and 55.5% at 0.1 mg/mL, but inhibitory rate at 1 mg/mL decreased up to 9.82% and 8.37%, respectively.
Of the samples tested, hot water extract of Portulaca oleracea extract had the highest inhibitory effect on fructosamine formation (by 45.60% at 1 mg/mL), followed by 60% EtOH extract of Aloe vera callus (35.75% at 1 mg/m) and 95% acetone extract of Aloe vera leaf (34.84 at 1 mg/mL).
AGE generation after the incubation of BSA with MGO during 7 days at 37 ℃ shows the effect on the middle stage of glycation. Of the 21 extracts tested, Aloe vera leaf extracts (chloroform: EtOH and 95% EtOH), Aloe arborescens extracts (hot water and 60% EtOH), and Aloe vera callus extract (60% EtOH) showed slight inhibitory effects. Portulaca oleracea and CBH extracts also inhibited slightly the MGO-mediated AGE formation in a concentration dependent manner. The Aloe vera leaf extract with chloroform: EtOH, 3:1,v/v) exhibited the highest inhibition of about 87%. AG and rutin as positive controls showed strong inhibitory effects in a dose-dependent manner, but (+) catechin and (−) epicatechin showed stronger inhibition at lower concentrations than at high concentrations. MGO reacts directly with proteins and contributes to AGE formation. In this study, the extracts inhibited MGO-mediated AGE formation. The inhibition of AGE formation may be due to the antioxidant activity or the MGO-trapping activity of the extracts. It has been recently shown that phenolic compounds are able to trap reactive dicarbonyl compounds, inhibiting the glycation reaction (Sang, et al., 2007). Our extracts contain phenolic compounds and also showed high antioxidant activity.
Fluorescence of AGEs, used as a readout for the effects on the late stage of glycation, was clearly decreased by Aloe vera leaf extracts (60% EtOH, chloroform, chloroform:EtOH, hexane, 95% and 60% acetone, and 95% EtOH) and by 60% EtOH extracts of Aloe arborescens leaves, Aloe vera callus, Portulaca oleracea and CBH. These extracts inhibited the formation of fluorescent AGEs in a dose-dependent manner. Among Aloe vera leaf extracts, the chloroform extract showed the strongest inhibition of AGE formation (99.9%), followed by the 95% acetone extract (92.77%) at concentration of 1 mg/ mL. These extracts exhibited higher anti-glycation activities than AG. Cold water, hot water and 60% EtOH CBH extracts had anti-glycation activities of 38.35%, 37.4% and 71.66%, respectively, at a concentration of 1 mg/mL. The positive controls (AG, rutin, (+) catechin and (−) epicatechin) reduced AGE fluorescence in a dose-dependent manner.
Recently, a number of studies have highlighted the benefits of using natural materials with anti-glycation and antioxidant properties. There are reports of plant-derived substances with AGE inhibitory effects. Ilex paraguariensis extracts and green tea extract have strong anti-glycation activities (40% at 0.02 mg/ mL and 15% at 0.005 mg/mL, respectively; Lunceford & Gugliucci, 2005; Nakagawa et al., 2002). In our study, chloroform and 95% acetone extracts of Aloe vera leaves had anti-glycation activities of 81.08% and 16.02%, respectively, at a concentration of 0.01 mg/mL. Cold water and hot water extracts of CBH have also showed inhibitory activities of 22.33% and 18.83%, respectively, at a concentration of 0.01 mg/mL. These data suggest that anti-glycation activities of Aloe vera leaf and CBH extracts were greater than those of Ilex paraguariensis extracts (Lunceford & Gugliucci, 2005). Plantago asiatica extract and wheat bran feruloyl oligosaccharides have been reported to have anti-glycation activities of 74.1% and 64.0%, respectively, at a concentration of 1 mg/mL (Choi et al., 2008; Wang et al., 2009), which was lower in comparison with the anti-glycation activities of Aloe vera leaf extracts (chloroform, 95% acetone) and CBH extract (60% EtOH) observed in our study.
Total phenolic and flavonoid contents in the extracts ranged from 58.11 to 493.85 and from 8.95 to 338.87 μg per 1 mg of powder, respectively (Table 2). The highest content of phenolics was found in the 95% acetone extract of Aloe vera leaves (49.06%, w/w), followed by the 60% EtOH extract of CBH (44.69%, w/w), chloroform: EtOH extract of Aloe vera leaves (30.35%, w/w) and 95% EtOH extract of Aloe vera leaves (20.86%, w/w). Most 60% EtOH extracts had higher phenolic contents than those of cold and hot water extracts.
The highest flavonoid content was found in the chloroform extract of Aloe vera leaves (33.33%, w/w) followed by the 60% EtOH extract of CBH (22.9%, w/w), chloroform: EtOH extract of Aloe vera leaves (19.21%, w/w), and 95% acetone extract of Aloe vera leaves (17.27%, w/w). These extracts had the highest total contents of phenolics and flavonoids.
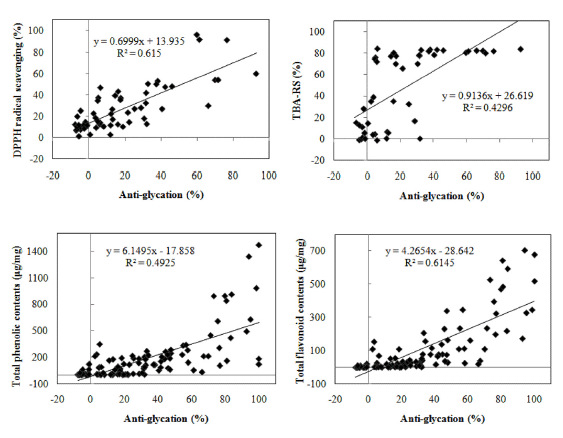
The relationship between the total flavonoid content, total phenolics content and anti-glycation activity of extracts was analyzed. As expected, the anti-glycation activity of the extracts correlated positively with the total contents of phenolics and flavonoids (Fig. 3; also see Table 3 for details). Glycation inhibition by Aloe arborescens extracts was significantly correlated with their DPPH radical scavenging activity (R 2 =0.946) and antioxidant activity by TBA-RS assay (R 2 =0.965). On the other hand, the correlation between the antiglycation activity of Portulaca oleracea and its antioxidant activity determined by the DPPH radical scavenging and TBARS assays was relatively weak. However, the overall correlation coefficient between the DPPH radical scavenging activity and anti-glycation activity was 0.659 (moderate correlation), which was higher than with TBA-RS. Moreover, the average correlation between the anti-glycation activity and flavonoid content (R 2 =0.615) revealed that the content of flavonoids was a major determinant of the anti-glycation activity of the extracts. This results is in line with those of Laurean et al. (2006) and Oya et al. (1997). The presence of high concentrations of flavonoids in the extracts could explain these properties.
Glycated proteins have been shown to provide stable active sites for catalyzing the formation of free radicals. These free radical reactions lead to the formation of fluorescent and nonfluorescent AGE adducts (McMorrough & McDowell, 1978; Wang et al., 2009).
It was found that free radical scavenging and high flavonoid contents are effective for glycation inhibition.
Conclusion
This present study was carried out to screen plants for their antiglycation activities. The anti-glycation capacities of samples were compared with that of AG (aminoguanidine) as a standard. Five kinds of samples, Aloe vera leaves, Aloe arborescens leaves, Aloe vera callus, Portulaca oleracea and cacao bean husk were extracted with hot water, cold water or 60% EtOH solution. Inhibition of protein glycation in vitro was examined by using the NBT assay, BSA-methylglyoxal assay and RNaseribose assay. Correlation between flavonoid or phenolics contents and the glycation inhibitory activity or the antioxidant potency of the extracts was analyzed.
The results of our study suggest that the extracts from Aloe vera leaves and CBH are rich in phenolic compounds, which probably contributes to their anti-glycation activities. This is the first report of anti-glycation activities of these samples. We suggest that extracts of Aloe vera leaves and CBH may potentially prevent AGE formation.